

Как наследуется гемофилия и современные способы лечения заболевания
Гемофилия
Заболевание из группы наследственных коагулопатий, обусловленное дефицитом факторов свертывания плазмы крови и характеризующееся повышенной склонностью к геморрагиям – кровотечениям и кровоизлияниям, связанным с нарушением целостности или проницаемости стенок сосудов.
Причины возникновения. Гены, обусловливающие развитие гемофилии, сцеплены с половой Х-хромосомой, поэтому заболевание наследуется по рецессивному признаку по женской линии. Наследственной гемофилией болеют практически исключительно лица мужского пола. Женщины являются проводниками (кондукторами, носителями) гена гемофилии, передающими заболевание части своих сыновей.
Врожденная гемофилия встречается почти у 70 % пациентов. В этом случае наследуется форма и тяжесть гемофилии. Около 30% наблюдений приходится на спорадические, спонтанные формы гемофилии, которые впоследствии становятся наследственными.
Риск передачи заболевания. У здорового мужчины и женщины-кондуктора с одинаковой вероятностью могут родиться как больные, так и здоровые сыновья. От брака мужчины, больного гемофилией, со здоровой женщиной рождаются здоровые сыновья или дочери-кондукторы. Описаны единичные случаи гемофилии у девочек, рожденных от матери-носителя и больного гемофилией отца.
Типы. В зависимости от дефицита того или иного фактора свертываемости крови, различают гемофилию А (классическую), В (болезнь Кристмаса), С и др.
Классическая гемофилия составляет подавляющее большинство (около 85%) случаев синдрома и связана с дефицитом VIII фактора свертывания (антигемофильного глобулина), приводящим к нарушению образования активной тромбокиназы.
При гемофилии В, составляющей 13% случаев заболевания, имеет место недостаток IX фактора (плазменного компонента тромбопластина, фактора Кристмаса), также участвующего в образовании активной тромбокиназы в I фазе свертывания крови.
Гемофилия С встречается с частотой 1-2% и обусловлена недостаточностью XI фактора свертывания крови (предшественника тромбопластина). На остальные разновидности гемофилии приходится менее 0,5% случаев; при этом может отмечаться дефицит различных плазменных факторов.
Симптомы. У новорожденных детей – длительное кровотечение из культи пуповины, подкожные гематомы, кефалогематомы.
У детей первого года жизни – длительные кровотечения в ходе прорезывания зубов, при оперативных вмешательствах. Острые края молочных зубов могут стать причиной прикусывания языка, губ, щек и кровотечений из слизистых оболочек полости рта. Однако в грудном возрасте гемофилия дебютирует редко в связи с тем, что материнском молоке содержится достаточное количество активной тромбокиназы.
Вероятность посттравматических кровотечений значительно возрастает, когда ребенок с гемофилией начинает вставать и ходить. Для детей после года характерны носовые кровотечения, подкожные и межмышечные гематомы, кровоизлияния в крупные суставы. Обострения геморрагического диатеза случаются после перенесенных инфекций (ОРВИ, ветрянки, краснухи, кори, гриппа и др.) вследствие нарушения проницаемости сосудов. Ввиду постоянных и длительных кровотечений у детей с гемофилией имеется анемия различной степени выраженности.
По степени убывания частоты кровоизлияния при гемофилии распределяются следующим образом:
гемартрозы (70—80%) – кровоизлияния в суставы;
гематомы (10-20%) – ограниченнное скопление крови при повреждении органов и тканей;
гематурия (14-20%) – наличие крови в моче;
желудочно-кишечные кровотечения (8%),
кровоизлияния в ЦНС (5%).
Лечение. В лечении гемофилии выделяют два направления – профилактические и «по требованию», в период проявлений геморрагического синдрома.
Профилактическое введение концентратов факторов свертывания крови показано пациентам с тяжелой формой гемофилии и проводится 2-3 раза в неделю для предупреждения развития гемофилической артропатии и прочих кровотечений. При развитии геморрагического синдрома требуются повторные трансфузии препарата. Дополнительно используются свежезамороженная плазма, эритромасса, гемостатики. Все инвазивные вмешательства у больных с гемофилией (наложение швов, удаление зубов, любые операции) проводятся под прикрытием гемостатической терапии.
При незначительных наружных кровотечениях (порезах, кровотечениях из полости носа и рта) используется гемостатическая губка, на рану накладывается давящая повязка, раны обрабатываются тромбином. При неосложненном кровоизлиянии ребенку необходим полный покой, холод, иммобилизация больного сустава гипсовой лонгетой, в дальнейшем – УВЧ, электрофорез, ЛФК, легкий массаж. Больным с гемофилией рекомендуется диета, обогащенная витаминами А, В, С, D, солями кальция и фосфора.
Профилактика. Проведение медико-генетического консультирования супружеских пар, имеющих отягощенный семейный анамнез по гемофилии. Дети, больные гемофилией, всегда должны иметь при себе специальный паспорт, где указан тип заболевания, группа крови и Rh-принадлежность.
Наблюдение. Пациентам показан охранительный режим, профилактика травм; диспансерное наблюдение педиатра, гематолога, детского стоматолога, детского ортопеда и др. специалистов; наблюдение в условиях специализированного гемофильного центра.
Гемофилия — что это за болезнь?
Как передается и каковы симптомы гемофилии?
Для большинства неосведомленных людей гемофилия – это так называемая царская болезнь, о ней знают только по истории: дескать, ею страдал царевич Алексей. Из-за недостатка знаний люди зачастую считают, что обычный народ болеть гемофилией не может. Есть мнение, что поражает она только древние роды. Но гемофилия – это наследственное заболевание, и его заполучить может любой ребенок, у которого предки имели такую болезнь.
Что такое гемофилия?
В народе болезнь называется «жидкая кровь». И действительно, ее состав патологичен, в связи с чем нарушена способность к свертыванию. Малейшая царапина – и кровотечение трудно остановить. Однако это внешние проявления. Гораздо более тяжелы внутренние, происходящие в суставах, желудке, почках. Кровоизлияния в них могут вызываться даже без постороннего воздействия и несут за собой опасные последствия.
За свертываемость крови отвечают двенадцать особых белков, которые должны присутствовать в крови в определенной концентрации. Заболевание гемофилия диагностируется в том случае, если один из этих белков отсутствует вовсе или наличествует в недостаточной концентрации.
В медицине выделяются три типа этого заболевания.
Гемофилия А. Вызывается отсутствием или недостатком фактора свертываемости VIII. Наиболее распространенный вид болезни составляет, согласно статистике, 85 процентов всех случаев заболевания. В среднем один младенец из 10 тысяч оказывается больным именно такой гемофилией.
Гемофилия В. При ней наблюдаются проблемы с фактором под номером IX. Отмечена как гораздо более редкая: риск ею заболеть в шесть раз ниже, чем в случае варианта А.
Гемофилия С. Недостает фактора номер XI. Эта разновидность уникальна: она свойственна и мужчинам, и женщинам. Стоит отметить, что в трети семей это заболевание встречается (или диагностируется) впервые, что становится ударом для неподготовленных родителей.
Почему возникает болезнь? Ее виновником становится врожденный ген гемофилии, который располагается на Х-хромосоме. Носителем его является женщина, причем сама она больной не является, разве что могут наблюдаться частые носовые кровотечения, слишком обильные менструации или более медленно заживающие мелкие ранки (например, после вырванного зуба). Ген является рецессивным, поэтому заболевают не все, у кого мать – носительница болезни. Обычно вероятность распределяется 50:50. Она повышается, если в семье болен еще и отец. Девочки же становятся носительницами гена в обязательном порядке.
Почему гемофилия – мужская болезнь. Как уже было сказано, ген гемофилии рецессивен и крепится на хромосому, обозначаемую как Х. У женщин таких хромосом две. Если одна поражена таким геном, она оказывается более слабой и подавляется второй, доминантной, вследствие чего девочка остается только носителем, через которого гемофилия передается, но сама остается здоровой. Вполне вероятно, что при зачатии обе Х-хромосомы могут содержать соответствующий ген. Однако при формировании плодом собственной кровеносной системы (а это происходит на четвертной неделе беременности) он становится нежизнеспособен, и происходит самопроизвольный аборт (выкидыш). Другое дело – мужчины. У них второй Х-хромосомы нет, ее заменяет Y. Доминантного «икса» нет, поэтому если рецессивный себя проявляет, то начинается именно протекание болезни, а не ее латентное состояние. Однако поскольку хромосом все же две, вероятность такого развития сюжета составляет ровно половину всех шансов.
Симптомы гемофилии. Они могут проявляться уже при рождении ребенка, если соответствующий фактор в организме у него практически отсутствует, а могут дать о себе знать только со временем, если наблюдается его недостаток. Кровоточивость при отсутствии явных причин. Нередко ребенок рождается с потеками крови из носа, глаз, пупка, и остановить кровотечение затруднительно. Гемофилия проявляет себя образованием больших отечных гематом от абсолютно незначительного воздействия (например, нажатия пальцем). Повторные кровотечения из вроде бы уже зажившей раны. Повышенная бытовая кровоточивость: носовая, из десен даже при чистке зубов. Кровоизлияния в суставы. Следы крови в моче и кале. Однако такие «приметы» необязательно свидетельствуют именно о гемофилии. К примеру, носовые кровотечения могут говорить о слабости стенок сосудов, кровь в моче – о почечных заболеваниях, а в кале – о язве. Поэтому обязательно проводятся дополнительные исследования.
Выявление гемофилии. Помимо изучения анамнеза больного и его осмотра самыми разными специалистами проводятся лабораторные анализы. В первую очередь определяется наличие в крови всех факторов свертывания и их концентрация. Устанавливается время, которое потребовалось для свертывания образца крови. Нередко эти анализы сопровождаются и исследованием ДНК. Для более точного диагноза может потребоваться определение: тромбинового времени; микста; протромбинового индекса; количества фибриногена. Иногда запрашиваются и более узкоспециализированные данные. Чем наиболее характерна гемофилия – это суставным кровотечением. Медицинское название – гемоартроз. Развивается он довольно быстро, хотя наиболее свойственен больным с тяжелыми формами гемофилии. У них кровоизлияния в суставы случаются без всякого внешнего воздействия, самопроизвольно. В легких формах для провоцирования гемоартроза требуется травма. Суставы поражаются в первую очередь те, которые испытывают нагрузки, то есть коленные, бедренные и пристопные. Вторые на очереди – плечевые, после них – локтевые. Первые симптомы гемоартроза проявляются уже у восьмилетних детей. Из-за суставных поражений большинство пациентов получают инвалидность.
Лечение гемофилии. К сожалению, гемофилия – это неизлечимая болезнь, которая сопровождает человека всю жизнь. Не придуман еще способ, которым можно заставить организм вырабатывать нужные белки, если он не умеет этого делать с рождения. Однако достижения современной медицины позволяют поддерживать организм на уровне, при котором больные гемофилией, особенно в не очень тяжелой форме, могут вести почти нормальное существование. Для предотвращения гематом и кровотечений требуется регулярное вливание растворов недостающих факторов свертывания. Их выделяют из крови людей-доноров и выращенных для донорства животных. Введение препаратов имеет постоянную основу в качестве профилактики и лечебную в случае предстоящей операции или травмы. Параллельно больные гемофилией должны постоянно проходить физиотерапию для поддержания работоспособности суставов. В случае слишком обширных, ставших опасными, гематом, хирурги делают операции по их удалению.
Что такое гемофилия?
Что такое гемофилия?
* Гемофилия, шире коагулопатия — заболевание крови, характеризующееся повышенной кровоточивостью, причиной которой является нарушение свертываемости крови.
* Нормальная свертываемость крови предотвращает и останавливает кровоизлияния в мышцы и суставы (гемартрозы и гематомы), а также кровотечения при порезах и царапинах, которые могут возникнуть при активной повседневной жизни любого человека.
* Свертывание крови — сложный физиологический процесс, в который вовлечено более десятка специальных белков — факторов свертывания крови, обозначаемых римскими цифрами от I до XIII. Дефицит фактора VIII называется гемофилией А, фактора IX — В. Дефицит или дефект (в зависимости от типа и подтипа) фактора Виллебранда называется болезнью Виллебранда. Существуют и более редкие коагулопатии, в частности, дефицит фактора XI — ранее называемый гемофилией С.
* Чем тяжелее гемофилия, тем раньше проявляются признаки кровоточивости. Неизбежные падения и ушибы, которые случаются у ребенка, начинающего ходить, могут вызвать синяки на коже и кровотечения из слизистых оболочек губ и языка.
* В возрасте 1-3 лет могут начаться поражения мышц и суставов, с болезненными припухлостями, ограничением движений рук и ног. Обширные гематомы, бывающие опасными, могут вызываться внутримышечными инъекциями. * При болезни Виллебранда наиболее частыми симптомами бывают кровотечения из слизистых носа.
* Иногда наличие диагноза вызывает сомнения, даже при повышенной кровоточивости у самого больного или наличии у других членов семьи гемофилии. Точная диагностика может быть сделана только измерением уровня соответствующего фактора свертывания крови. Такие анализы проводятся в специализированных лабораториях гематологических центров. *
* Гемофилия А и В поражает почти исключительно мужчин, а передается по женской линии. По статистике ВОЗ примерно 1 младенец мужского пола из 5000 рождается с гемофилией А, вне зависимости от национальной или расовой принадлежности. Приблизительно одна треть больных гемофилией А не имела у предшествующих поколений подобных расстройств.
* Наследование болезни Виллебранда и гипопроконвертинемии не сцеплено с полом.
* Лечение гемофилии. * В прошлом, когда лечение гемофилии производилось устаревшими, не защищенными от вирусов препаратами, инвалидизация больных с тяжелой формой заболевания достигала 90%, а инфицированность гепатитом С — 95%.
* Сейчас гемофилию А и В лечат концентратами соответствующих факторов свертывания крови. Комплекты поставки препарата включают в себя лиофилизированный (высушенный) фактор свертывания, воду для растворения, а также приспособления для внутривенных инъекций. Таким образом, имеется возможность для лечения пациента вне медицинского учреждений. Этот тип лечения называется домашним.
* Гематологи с раннего детства при тяжелой форме заболевания рекомендуют больным гемофилией профилактическое лечение. При таком лечении, инъекции фактора производятся не по факту кровоизлияния (кровотечения), а вне зависимости от него, по специальному расписанию, составленному врачом-гематологом.
* В последние годы в России удалось достичь уровня обеспечения пациентов с гемофилией А и В на уровне развитых стран Запада. Благодаря этому, новое поколение больных гемофилией минимально инвалидизировано. Препараты проходят вирусную инактивацию и шанс заразиться известными на данный момент вирусными инфекциями, передаваемыми через кровь, в современных условиях стремится к нулю.
Гемофилия
Гемофилия – наследственная патология гомеостаза, проявляющаяся в нарушении свертывания крови на фоне сниженного синтеза необходимых веществ организмом пациента (факторов VII, IX, XI). Страдающие от заболевания лица регулярно находят на своем теле гематомы или сталкиваются с гемартрозами, внутренними кровотечениями на фоне травм или хирургических вмешательств. Лечение гемофилии у детей строится на заместительной терапии, которую необходимо получать на протяжении всей жизни.
Причины развития патологии

Механизм и тип наследования гемофилии изучены достаточно подробно. Гены, провоцирующие недостаточную выработку факторов свертывания крови, сцеплены с Х-хромосомой. Патология наследуется по рецессивному признаку по женской линии. Наследственно обусловленная патология встречается исключительно у мальчиков.
Сыновья здорового мужчины и женщины-носительницы патологического гена с одинаковой вероятностью могут родиться без признаков гемофилии или с ними. Мужчина, страдающий от нарушений свертывания крови, сможет зачать здоровых детей с женщиной, не являющейся носительницей измененного гена.
Медицине известны единичные случаи обнаружения гемофилии у женщин. Их матери были носительницами мутировавшего гена, а отцы страдали от недостаточной выработки факторов свертывания крови. Причинами гемофилии в подобных случаях становится соединение рецессивного и доминантного генов.
Классификация заболевания
Виды гемофилии выделяются на основании дефицита определенного фактора свертываемости крови в организме пациента. Основные сведения о типах заболевания представлены в таблице.
Диагностируется у 84-86% людей, страдающих от рассматриваемой патологии. Вызван дефицитом VIII фактора свертываемости (антигемофильного глобулина).
Занимает долю в 12-13% от клинически диагностируемых случаев гемофилии. Развивается на фоне дефицита IX фактора свертываемости (тромбопластина)
Встречается не чаще 1-2%. Становится следствием недостаточной выработки XI фактора свертывания крови.
Около 0,5% выявляемых случаев гемофилии различной этиологии относятся к иным типам – V, VII, X и другим.
Симптомы гемофилии
Первые признаки патологии можно обнаружить у новорожденных детей. Так, на вероятность подтверждения диагноза «гемофилия» у ребенка укажут длительное кровотечение из пуповины и многочисленные подкожные гематомы. Обильное истечение крови у младенцев в момент прорезывания зубов становится не менее тревожным фактором. Но в период лактации ребенок получает достаточное количество тромбокиназы с молоком матери.
Посттравматические кровотечения характерны для детей, научившихся ползать и ходить. К носовым кровотечения добавляются межмышечные гематомы и кровоизлияния в суставы (гемартрозы). На фоне этого развивается анемия.
С возрастом дети могут столкнуться с истечением крови из органов ЖКТ. Причиной этого становятся травмы или медицинские манипуляции. Наибольшую опасность представляют кровотечения из носоглотки и зева. На их фоне может развиться обструкция дыхательных путей.
Диагностика патологии
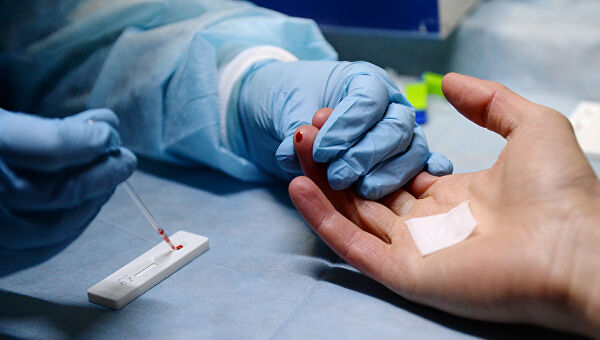
Диагностические процедуры выполняются врачами нескольких специализаций: неонатологом, педиатром, генетиком и гематологом. При сопутствующих патологиях могут потребоваться консультации с гастроэнтерологом, ортопедом, отоларингологом и неврологом.
Семейным парам из группы риска следует посетить врача до зачатия ребенка. Молекулярно-генетическое исследование биоматериалов будущих родителей позволит учесть риск рождения ребенка с гемофилией. После зачатия возможно выполнение пренатальных скринингов. Их результаты подтвердят или опровергнут факт наследования гемофилии ребенком.
Неонатальные тесты, выполняемые в первые дни жизни малыша, оказываются не менее эффективности. Коагулограмма дает неонатологу исчерпывающие сведения о продуцировании факторов свертываемости организмом новорожденного.
При гемартрозах ребенку назначается рентгенографическое исследование суставов. Ультразвуковая диагностика выполняется при обнаружении признаков внутренних кровотечений и забрюшинных гематом.
Лечение заболевания
Терапевтические мероприятия на фоне лечения гемофилии относятся к одной из двух групп – профилактическим или симптоматическим. В первом случае пациент с тяжелой формой патологии получает регулярные дозы концентратов факторов свертывания крови. Их присутствие снижает риск развития артропатии и внутренних кровотечений. При планировании оперативных вмешательств (включая удаление зубов) пациент получает повторные трансфузии препаратов.
Симптоматические процедуры выполняются при порезах и наружных кровотечениях. Врачи рекомендуют использовать гемостатическую губку и накладывать давящую повязку. Мелкие раны следует обрабатывать тромбином.
Прогноз
Заместительная терапия позволяет организму пациента выработать достаточное количество антител, которые блокируют прокоагулянтную активность. Дополнительными мерами поддержания мальчиков или мужчин становится плазмаферез и прием иммунодепрессантов. На фоне регулярных переливаний крови и плазмы пациенты могут столкнуться в ВИЧ, гепатитом, герпесом или цитомегаловирусом.
Легкая степень гемофилии не оказывает существенного влияния на качество и продолжительности жизни пациентов. При тяжелых формах патологии прогноз выживаемости существенно ухудшается. Клинические рекомендации при гемофилии предусматривают регулярные консультации пациентов с гематологом и контроль показателей свертываемости крови посредством лабораторных тестов.
Статистика
Гемофилия относится к наследственным коагулопатиям – причина развития заболевания обусловлена особенностями генетического материала матери и отца ребенка. В Москве патология типов A и B выявляется с частотой 1 случай на 10000 и 50000 новорожденных мальчиков соответственно. Симптомы заболевания диагностируются в раннем возрасте в ходе осмотра младенца неонатологом или педиатром.
Вопросы и ответы
Можно ли полностью излечиться от гемофилии?
Медикаментозная терапия и манипуляции по переливанию крови или плазмы купируют острые синдромы патологии. Иногда пациентам назначается длительный прием препаратов, подобранных с учетом данных анамнеза. Но полное устранение симптомов гемофилии по-прежнему остается невозможным.
Несет ли гемофилия угрозу жизни ребенка?
При своевременном обращении родителей за медицинской консультацией малыш окажется вне опасности. Быстрая постановка правильного диагноза и начало лечения позволят ребенку не ограничивать себя в двигательной активности и играх.
Может ли мальчик, болеющий гемофилией, передать ее своим сыновьям?
Риск рождения детей с гемофилией от отца, испытывающего проблемы со свертыванием крови, минимален. Заболевание будет унаследовано сыновьями только в том случае, если их мать входит в число носителей измененного гена.
Существует ли эффективный механизм профилактики гемофилии?
Нет, такого механизма не существует. Но будущие родители могут пройти скрининги, которые определяет вероятность рождения страдающего от недостаточной свертываемости крови ребенка.
Фенилкетонурия
Фенилкетонурия – наследственное нарушение аминокислотного обмена, обусловленное недостаточностью печеночных ферментов, участвующих в метаболизме фенилаланина до тирозина. Ранними признаками фенилкетонурии служат рвота, вялость или гиперактивность, запах плесени от мочи и кожи, задержка психомоторного развития; типичные поздние признаки включают олигофрению, отставание в физическом развитии, судороги, экзематозные изменения кожи и др. Скрининг новорожденных на фенилкетонурию проводится еще в родильном доме; последующая диагностика включает молекулярно-генетическое тестирование, определение концентрации фенилаланина в крови, биохимический анализ мочи, ЭЭГ, МРТ головного мозга. Лечение фенилкетонурии заключается в соблюдении специальной диеты.
Общие сведения
Фенилкетонурия (болезнь Феллинга, фенилпировиноградная олигофрения) – врожденная, генетически обусловленная патология, характеризующаяся нарушением гидроксилирования фенилаланина, накоплением аминокислоты и ее метаболитов в физиологических жидкостях и тканях с последующим тяжелым поражением ЦНС. Фенилкетонурия впервые описана А. Феллингом в 1934 г.; встречается с частотой 1 случай на 10 000 новорожденных. В неонатальном периоде фенилкетонурия не имеет клинических проявлений, однако поступление фенилаланина с пищей вызывает манифестацию заболевания уже в первом полугодии жизни, а в дальнейшем приводит к тяжелым нарушениям развития ребенка. Именно поэтому пресимптоматическое выявление фенилкетонурии у новорожденных является важнейшей задачей неонатологии, педиатрии и генетики.
Причины фенилкетонурии
Фенилкетонурия является заболеванием с аутосомно-рецессивным характером наследования. Это означает, что для развития клинических признаков фенилкетонурии ребенок должен унаследовать по одной дефектной копии гена от обоих родителей, являющихся гетерозиготными носителями мутантного гена.
Чаще всего к развитию фенилкетонурии приводит мутация гена, кодирующего фермент фенилаланин-4-гидроксилазу и расположенного на длинном плече 12 хромосомы (локус12q22-q24.1). Это, так называемая, классическая фенилкетонурия I типа, составляющая 98% всех случаев заболевания. Гиперфенилаланинемия может достигать 30 мг% и выше. При отсутствии лечения данный вариант фенилкетонурии сопровождается глубокой умственной отсталостью.
Кроме классической формы, различают атипичные варианты фенилкетонурии, протекающие с той же клинической симптоматикой, но не поддающиеся коррекции диетотерапией. К ним относятся фенилкетонурия II типа (недостаточность дегидроптеринредуктазы), фенилкетонурия III типа (дефицит тетрагидробиоптерина) и другие, более редкие варианты.
Вероятность рождения ребенка, больного фенилкетонурией, повышается при заключении близкородственных браков.
Патогенез фенилкетонурии
В основе классической формы фенилкетонурии лежит недостаточность фермента фенилаланин-4-гидроксилазы, участвующего в конверсии фенилаланина в тирозин в митохондриях гепатоцитов. В свою очередь, производный тирозина – тирамин является исходным продуктом для синтеза катехоламинов (адреналина и норадреналина), а дийодтирозин – для образования тироксина. Кроме этого, результатом метаболизма фенилаланина служит образование пигмента меланина.
Наследственная недостаточность фермента фенилалаиин-4-гидроксилазы при фенилкетонурии приводит к нарушению окисления фенилаланина, поступающего с пищей, в результате чего его концентрация в крови (фенилаланинемия) и спинномозговой жидкости значительно возрастает, а уровень тирозина соответственно падает. Избыточное содержание фенилаланина устраняется путем повышенной экскреции с мочой его метаболитов — фенилпировиноградной, фенилмолочной и фенилуксусной кислот.
Нарушение обмена аминокислот сопровождается нарушением миелинизации нервных волокон, снижением образования нейромедиаторов (дофамина, серотонина и др.), запускающими патогенетические механизмы задержки умственного развития и прогредиентное слабоумие.
Симптомы фенилкетонурии
Новорожденные с фенилкетонурией не имеют клинических признаков заболевания. Обычно манифестация фенилкетонурии у детей происходит в возрасте 2-6 месяцев. С началом кормления в организм ребенка начинает поступать белок грудного молока либо его заменителей, что приводит к развитию первых, неспецифических симптомов – вялости, иногда – беспокойства и гипервозбудимости, срыгивания, мышечной дистонии, судорожного синдрома. Одним из ранних патогномоничных признаков фенилкетонурии служит упорная рвота, которая нередко ошибочно расценивается как проявление пилоростеноза.
Ко второму полугодию становится заметным отставание ребенка в психомоторном развитии. Ребенок становится менее активным, безучастным, перестает узнавать близких, не пытается садиться и вставать на ножки. Аномальный состав мочи и пота обусловливают характерный «мышиный» запах (запах плесени), исходящий от тела. Часто наблюдается шелушение кожи, дерматиты, экзема, склеродермия.
У детей с фенилкетонурией, не получающих лечения, выявляется микроцефалия, прогнатия, позднее (после 1,5 лет) прорезывание зубов, гипоплазия эмали. Отмечается задержка речевого развития, а к 3-4 годам выявляется глубокая олигофрения (идиотия) и практически полное отсутствие речи.
Дети с фенилкетонурией имеют диспластическое телосложение, нередко — врожденные пороки сердца, вегетативные дисфункции (потливость, акроцианоз, артериальную гипотонию), страдают запорами. К фенотипическим особенностям детей, страдающих фенилкетонурией, следует отнести светлую кожу, глаза и волосы. Для ребенка с фенилкетонурией характерны специфическая поза «портного» (согнутые в суставах верхние и нижние конечности), тремор рук, шаткая, семенящая походка, гиперкинезы.
Клинические проявления фенилкетонурии II типа характеризуются тяжелой степенью умственной отсталости, повышенной возбудимостью, судорогами, спастическим тетрапарезом, сухожильной гиперрефлексией. Прогрессирование заболевание может приводить к гибели ребенка в возрасте 2-З лет.
При фенилкетонури III типа развивается триада признаков: микроцефалия, олигофрения, спастический тетрапарез.
В настоящее время диагностика фенилкетонурии (а также галактоземии, врожденного гипотиреоза, адрено-генитального синдрома и муковисцидоза) входит в программу неонатального скрининга, осуществляемого всем новорожденным.
Скрининг-тест проводится на 3-5 день жизни доношенного и 7 день жизни недоношенного ребенка путем забора образца капиллярной крови на специальный бумажный бланк. При обнаружении гиперфенилаланемии более 2,2 мг% ребенка направляют к детскому генетику для повторного обследования.
Для подтверждения диагноза фенилкетонурии проверяется концентрация фенилаланина и тирозина в крови, определяют активность печеночных ферментов (фенилаланингидроксилазы), выполняется биохимическое исследование мочи (определение кетоновых кислот), метаболитов катехоламинов в моче и др. Дополнительно проводится ЭЭГ и МРТ головного мозга, осмотр ребенка детским неврологом.
Генетический дефект при фенилкетонурии может быть обнаружен еще на этапе беременности в ходе инвазивной пренатальной диагностики плода (хорионбиопсии, амниоцентеза, кордоцентеза).
Дифференциальный диагноз фенилкетонурии проводят с внутричерепной родовой травмой новорожденных, внутриутробными инфекциями, другими нарушениями обмена аминокислот.
Лечение фенилкетонурии
Основополагающим фактором в лечении фенилкетонурии является соблюдение диеты, ограничивающей поступление белка в организм. Лечение рекомендуется начинать при концентрации фенилаланина >6 мг%. Для грудных детей разработаны специальные смеси — Афенилак, Лофенилак; для детей старше 1 года – Тетрафен, Фенил-фри; старше 8 лет — Максамум-ХР и др. Основу диеты составляют низкобелковые продукты — фрукты, овощи, соки, белковые гидролизаты и аминокислотные смеси. Расширение диеты возможно после 18 лет в связи с возрастанием толерантности к фенилаланину. В соответствии с российским законодательством обеспечение лиц, страдающих фенилкетонурией, лечебным питанием, должна осуществляться бесплатно.
Больным назначается прием минеральных соединений, витаминов группы В и др.; по показаниям — ноотропные средства, антиконвульсанты. В комплексной терапии фенилкетонурии широко используется общий массаж, ЛФК, иглорефлексотерапия.
Дети, страдающие фенилкетонурией, находятся под наблюдением участкового педиатра и психоневролога; нередко нуждаются в помощи логопеда и дефектолога. Необходим тщательный мониторинг нервно-психического статуса детей, контроль уровня фенилаланина в крови и показателей электроэнцефалограммы.
Атипичные формы фенилкетонурии, не поддающиеся лечению диетой, требуют назначения гепатопротекторов, противосудорожных средств, заместительной терапии леводопой, 5-гидрокситриптофаном.
Прогноз и профилактика фенилкетонурии
Проведения массового скрининга на фенилкетонурию в неонатальном периоде позволяет организовать раннюю диетотерапию и предотвратить тяжелые церебральные повреждения, нарушения функции печени. При раннем назначении элиминационной диеты при классической фенилкетонурии прогноз развития детей хороший. При поздно начатом лечении прогноз в отношении умственного развития неблагоприятный.
Профилактика осложнений фенилкетонурии заключается в проведении массового скрининга новорожденных, раннего назначения и длительного соблюдения диетического питания.
С целью оценки риска рождения ребенка с фенилкетонурией предварительное генетическое консультирование должны пройти супружеские пары, уже имеющие больного ребенка, состоящие в кровнородственном браке, имеющие родственников с данным заболеванием. Женщины с фенилкетонурией, планирующие беременность, должны соблюдать строгую диету до зачатия и во время беременности для исключения повышения уровня фенилаланина и его метаболитов и нарушения развития генетически здорового плода. Риск рождения ребенка с фенилкетонурией у родителей-носителей дефектного гена, составляет 1:4.
Гемофилия
Гемофилия — заболевание крови, которое относится к группе наследственных коагулопатий.
Распространенность патологии в мире 1 случай на 10 тыс. мужского населения. В народе популярно мнение, что больной с гемофилией может внезапно скончаться от тяжелого кровотечения, которое невозможно остановить. Это не соответствует действительности и порождено слухами. Однако риск летального исхода от геморрагического инсульта, действительно, превышает средние показатели, поскольку уязвимость сосудов мозга велика.
Причины
Причины гемофилии достаточно изучены. Обнаружены характерные изменения одного гена в Х-хромосоме. Установлено, что именно этот участок ответственен за производство необходимых факторов свертывания, специфических белковых соединений.
70% случаев гемофилии считаются наследственными. Генетические изменения наследуются по рецессивному типу (доминантный тип указывает на болезнь обоих родителей и обязательную передачу детям). Роль женщины как носителя мутации сводится к передаче заболевания сыновьям. Рожденная девочка может быть носительницей, но сама не болеть.
Вероятность рождения больного мальчика у пары, состоящей из здорового мужчины и матери-носительницы, составляет 50:50. Редко встречаются случаи заболевания у девочек, рожденных от больного гемофилией отца и здоровой матери. Обычно от подобного брака можно ожидать здоровых сыновей или дочек-носительниц измененного гена.
Гемофилия у женщин (девочек) – очень редкое явление. Для этого ребенок должен родиться от больного гемофилией отца и матери-носительницы. В мировой практике описано всего 60 аналогичных заболеваний. Успехи современной медицины по продлению жизни пациентов предполагают рост выявления больных девочек в ближайшие годы.
Почему гемофилию называют «царской болезнью»?
Появлению названия «царская болезнь» патология обязана британской королеве Виктории. Именно она послужила родоначальником измененного гена у своих детей и внуков. При этом проверены исторические свидетельства о возможности отцовства Виктории и связей матери.
Клинические проявления гемофилии обнаружены у сына Виктории Леопольда, герцога Олбани. Далее наследование шло по рецессивному признаку через дочерей и внучек. Одним из факторов, усиливающих вероятность заболевания, считаются близкородственные браки между кузенами в королевских семьях Европы.
Таким образом, мутирующий ген достался через императрицу Александру царевичу Алексею Николаевичу, сыну последнего российского монарха.
Виды гемофилии
Тип А — вызван недостатком антигемофильного глобулина (фактора VIII), нарушающего синтез тромбокиназы. Наиболее типичная форма. Ей болеют до 85% пациентов. Тяжесть обусловлена кровотечениями при травмах, операциях, встречающихся на фоне снижения концентрации фактора до 5–20% от нормального уровня.
Тип В (болезнь Кристмаса) — связан с недостатком синтеза фактора IX, ответственного за создание вторичной коагулирующей пробки в сосуде и синтез тромбокиназы, болеют до 15% пациентов.
Классификация по тяжести течения
На тяжесть течения заболевания влияет степень недостатка перечисленных факторов, вызывающих потерю способности крови к свертываемости. Различают степени:
- легкую — концентрация фактора составляет 6% и выше, выявляется обычно в школьном возрасте, кровотечения редкие, мало интенсивные, возникают при травмах, в ходе операции, при экстракции зуба;
- среднетяжелую — уровень факторов колеблется от 1 до 5% от нормы, обычно симптомы гемофилии выявляются в дошкольные годы, характерны умеренно выраженные геморрагические проявления (кровоизлияния в суставные сумки, мышцы), в анализе мочи обнаруживают эритроциты;
- тяжелую — уровень дефицита ниже 1%, уже в раннем детстве развиваются выраженные геморрагические проявления, у новорожденного определяют распространенные гематомы на голове, усиленное кровотечение из пуповины, черный стул (мелену), гематомы возникают без связи с травмированием, характерна кровоточивость десен при прорезывании зубов, смене молочных на коренные, возможны кровоизлияния во внутренние органы.
Клинические признаки
Для симптомов гемофилии типов А и В типичны:
- различные гематомы (в мышцах, под кожей, под мышечными фасциями, в забрюшинном пространстве) чаще связаны с небольшим ушибом, имеются у каждого пятого пациента, отличаются обширной зоной распространения, если сдавливается внутренний нервный ствол или артерия, то появляются сильные боли;
- кровотечения — из порезов, лунки удаленного зуба, десен, после хирургических вмешательств, травм, характерен отсроченный характер (кровотечение возникает не сразу после травмы, а спустя 6–8 часов);
- возникновение гемартрозов (70–80% случаев);
- гематурия — почечное кровотечение, обнаруживаемое по примеси крови в моче (от 14 до 20% случаев), возникает самопроизвольно или после ушиба поясницы, может сопровождаться приступами почечной колики, приводит к гидронефрозу, хроническому пиелонефриту;
- кишечное кровотечение — проявляется у 8% больных тяжелой формой гемофилии в виде «черного» жидкого стула, резкого головокружения, слабости, может провоцироваться приемом лекарственных средств (стероиды, аналгетики);
- кровоизлияние в брыжейку — вызывает приступ острых болей в животе, невозможно отличить от аппендицита и перитонита, иногда сходно с клиникой острой кишечной непроходимости;
- геморрагические инсульты — сопровождают до 5% заболевших, чаще возникают в молодом возрасте, клиника зависит от локализации очага.
Диагностические признаки
Определяют измененные показатели коагулограммы:
- увеличение активированного частичного тромбопластинового времени (АЧТВ);
- снижение количества факторов свёртывания крови
Лечение
Лечение гемофилии заключается в заместительной терапии. Избавить больного от патологии невозможно, приходится с помощью лекарственных препаратов добавлять или вводить в организм пациента дефицитные факторы свертывания.
Необходимая дозировка зависит от степени недостаточности, вида и тяжести течения заболевания. Потребность в повышении дозы возникает при выработке антител на замещающие белковые комплексы.
Выделяют 2 направления терапии гемофилии:
- профилактика в периоды без признаков геморрагического синдрома ;
- лечение в период геморрагических проявлений.
Профилактическое лечение гемофилии сводится к введению факторов свертывания в поддерживающей дозировке 2–3 раза в неделю. Все хирургические вмешательства, включая удаление зубов, проводятся «под прикрытием» добавочной дозировки факторов. Это позволяет предупредить развитие поражения суставов и других кровоизлияний.
Лечение в период геморрагических проявлений сводится к введению в организм пациента дефицитных факторов свертывания
Ученые-генетики разрабатывают меры воздействия на измененный геном больного человека. Пока проводятся только эксперименты на животных. Результативность получена при использовании ферментативной базы аденоассоциированных вирусов. Как оказалось, они способны удалять мутирующую часть гена гемофилии и заменять ее на здоровую.
Прогноз жизни для нынешних гемофиликов благоприятен!
Главный внештатный специалист по детской гематологии главного управления по здравоохранению Брестского облисполкома

