Особенности течения и лечения галактоземии у детей
Генетические заболевания
Галактоземия наследуется по аутосомно-рецессивному типу, то есть ребенок будет больной только в том случае, если унаследует две копии дефектного гена (по одной от каждого родителя). Гетерозиготные лица считаются носителями болезни, ведь они наследуют один нормальный ген и один дефектный ген. Но, у носителей, как правило, тоже проявляются некоторые симптомы галактоземии, но, конечно, в очень умеренной форме.
Типы
Галактоза превращается в глюкозу под действием трех ферментов. Этот процесс известен под названием метаболический путь Лелуара. Соответственно, к количеству задействованных в процессе ферментов известно 3 типа галактоземии (1, 2 и 3 типа):
Накопление галактозы приводит к активации определенных путей ее утилизации:
1. Обновление к галактиолу
У людей, больных галактоземией, накопление галактозы становится субстратом для ферментов, катализирующих процесс углеводного обмена (полиоловый путь). Первой реакцией в этой последовательности является превращение альдозы, (типа углеводов, содержащих альдегидную группу, к которому относится и галактоза) в сорбитол (углевод, который относится к классу многоатомных спиртов). Известно, что существует фермент, который называется альдозо-редуктаза, который является катализатором полиолового пути. Этот фермент превращает галактозу в ее спиртовую форму, галактиол. Однако галактиол не переходит на следующий этап полиолового пути с помощью фермента полиол-дегидрогеназы. Таким образом, галактиол не выполняет никаких функций и просто накапливается в тканях организма и выводится с мочой больных галактоземией. Накопление галактиола приводит к появлению многих осложнений, которые возникают у больных галактоземией. Его высокая концентрация была обнаружена у больных с классической галактоземией (дефицит GALT), а также у больных на дефицит галактокиназы и дефицит эпимеразы.
2. Окисление до галактоната
Галактоза, которая накапливается в организме больных галактоземией может принимать участие и в другой, (альтернативной к предыдущей) реакции, окислении до галактоната (галактоновой кислоты).
Подробный механизм образования галактоната, на сегодня не известен. Однако ученые предполагают, что под действием фермента галактодегидрогеназы происходит превращение галактозы в галактонолактон, который затем спонтанно или под действием определенных ферментов превращается в галактонат. После образования галактоната может начаться пентозофосфатный путь. Таким образом, окисление до галактоната, служит альтернативной формой метаболизма галактозы. То есть, этот процесс можно назвать менее вредным для организма, чем накопление галактиола.
Диагностика
В США дети регулярно проверяются на наличие галактоземии, поэтому в подавляющем большинстве случаев, если устанавливается диагноз — галактоземия, то это происходит в детском возрасте, что очень важно для дальнейшего лечения. У младенцев, пораженных галактоземией, обычно присутствуют симптомы:
- летаргии,
- у них возникают приступы рвоты, поноса,
- они очень медленно развиваются (физически),
- кроме того у них наблюдается желтуха.
Тестирование на наличие (или отсутствие галактоземии) проводится также с помощью анализа крови (кровь берется из пяты ребенка) или мочи. При исследовании этих анализов, проверяется наличие трех ферментов, необходимых для расщепления лактозы, (который содержится в молоке и молочных продуктах) на глюкозу и галактозу (которые организм использует для образования энергии). У человека, больного галактоземией, как правило, нет какого-либо из этих ферментов, и поэтому в крови или моче наблюдается высокий уровень галактозы.
Галактоземию, обычно обнаруживают во время скрининга новорожденных. Галактоземия может серьезно повлиять на этих детей, приведя при этом к необратимым последствиям, или даже смерти больного ребенка на протяжении первых дней жизни. Именно поэтому, важно, чтобы новорожденный ребенок прошел обследование на наличие любых метаболических отклонений. То есть, галактоземия может быть обнаружена еще до первого приема грудного молока или молочных смесей, содержащих галактозу.
Выявление расстройства не зависит от того употреблял ребенок белок или галактозу, именно поэтому наличие заболевания должно быть видно сразу после проведения анализов. Однако, полученные образцы нужно исследовать очень быстро, ведь, как правило ферменты разрушаются при хранении и от воздействия высоких температур. Однако сегодня во время скрининга новорожденных достаточно точно определяется наличие (или отсутствие) галактоземии. Для этого используют два известных скрининг тесты — тест Бутлера и тест Хилла (Beutler’s test and the Hill test). Недавно появился еще один тест — «Florida test», который также достаточно точно определяет наличие заболевания.
Процесс исследования благодаря использованию теста Бутлера позволяет определить уровень ферментов у новорожденных. То есть, употребление смесей или грудного молока не влияет на результаты исследования и поэтому достаточно точно выявляет галактоземию, несмотря на то попадала ли в организм ребенка галактоза или нет.
Лечение
Единственным лечением при классической галактоземии является строгое воздержание от употребления галактозы и лактозы. Однако, даже при условии ранней диагностики и соблюдении диеты, со временем могут возникнуть следующие осложнения: нарушение речи, трудности при обучении, неврологические нарушения (например, тремор и т.д.). Однако эти симптомы не проявляются у больных галактоземией Дуарте, и вообще, этим пациентам часто не нужно ограничивать свой рацион. Младенцы с классической галактоземией не могут питаться грудным молоком, из-за присутствия там лактозы, поэтому, как правило, они употребляют смеси на основе соевого молока.
Галактоземию иногда путают с непереносимостью лактозы, но при галактоземии вероятность возникновения различных нарушений — выше. Ведь проблемы с усвоением организмом лактозы возникают или наследуются, из-за дефицита фермента лактазы. Проявлениями этого заболевания являются боли в животе после приема молочных продуктов, но нет никаких долгосрочных последствий воздействия болезни. В то время когда при галактоземии, употребление человеком продуктов, содержащих галактозу, может привести к необратимому повреждению органов.
Долгосрочные последствия галактоземии:
Галактоземия

Галактоземия — наследственная ферментопатия, характеризующаяся нарушением нормального процесса углеводного обмена, а именно – метаболизма галактозы. Признаками галактоземии являются непереносимость грудного молока и молочных смесей, рвота, анорексия, гипотрофия, желтуха, цирроз печени, спленомегалия, отеки, катаракта, задержка психомоторного развития. Скрининг на галактоземию проводится всем новорожденным; дополнительное обследование включает определение уровня галактозы в крови и моче, проведение нагрузочных проб с галактозой и глюкозой, генетическое тестирование, УЗИ брюшной полости, ЭЭГ и др. Основу терапии галактоземии составляет безлактозная диета, назначаемая с первых дней жизни ребенка.
- Причины галактоземии
- Симптомы галактоземии
- Диагностика галактоземии
- Лечение галактоземии
- Прогноз и профилактика галактоземии
- Цены на лечение
Общие сведения
Галактоземия – наследственная патология обмена веществ, обусловленная недостаточностью активности ферментов, принимающих участие в метаболизме галактозы. Неспособность организма утилизировать галактозу приводит к тяжелым поражениям пищеварительной, зрительной и нервной системы детей в самом раннем возрасте. В педиатрии и генетике галактоземия относится к редким генетическим заболеваниям, встречающимся с частотой один случай на 10 000 — 50 000 новорожденных.
Впервые клиника галактоземии была описана в 1908 году у ребенка, страдавшего сильным истощением, гепато- и спленомегалией, галактозурией; при этом заболевание исчезло сразу после отмены молочного питания. Позднее, в 1956 г. ученый Герман Келкер определил, что в основе заболевания лежит нарушение метаболизма галактозы.
Причины галактоземии
Галактоземия является врожденной патологией, наследуемой по аутосомно-рецессивному типу, т. е. заболевание проявляется только в том случае, если ребенок наследует две копии дефектного гена от каждого из родителей. Лица, гетерозиготные по мутантному гену, являются носителями заболевания, однако у них тоже могут развиваться отдельные признаки галактоземии в легкой степени.
Превращение галактозы в глюкозу (метаболический путь Лелуара) происходит при участии 3-х ферментов: галактоза-1-фосфатуридилтрансферазы (GALT), галактокиназы (GALK) и уридиндифосфат-галактозо-4-эпимеразы (GALE). В соответствии с дефицитом этих ферментов различают 1 (классический вариант), 2 и 3 тип галактоземии.
Выделение трех типов галактоземии не совпадает с порядком действия ферментов в процессе метаболического пути Лелуара. Галактоза поступает в организм с пищей, а также образуется в кишечнике в процессе гидролиза дисахарида лактозы. Путь метаболизма галактозы начинается с ее превращения под действием фермента GALK в галактозо-1-фосфат. Затем при участии фермента GALT галактозо-1-фосфат преобразуется в УДФ-галактозу (уридилдифосфогалактозу). После этого с помощью GALE метаболит превращается в УДФ – глюкозу (уридилдифосфоглюкозу).
При недостаточности одного из названных ферментов (GALK, GALT или GALE) концентрация галактозы в крови значительно повышается, в организме накапливаются промежуточные метаболиты галактозы, которые вызывают токсическое поражение различных органов: ЦНС, печени, почек, селезенки, кишечника, глаз и др. Нарушение метаболизма галактозы и составляет суть галактоземии. Наиболее часто в клинической практике встречается классический (1 тип) галактоземии, обусловленный дефектом фермента GALT и нарушением его активности. Ген, кодирующий синтез галактоза-1-фосфатуридилтрансферазы, находится в околоцентромерном участке 2-ой хромосомы.
Симптомы галактоземии
По тяжести клинического течения выделяют тяжелую, среднюю и легкую степени галактоземии.
Первые клинические признаки галактоземии тяжелой степени развиваются очень рано, в первые дни жизни ребенка. Вскоре после кормления новорожденного грудным молоком или молочной смесью возникает рвота и расстройство стула (водянистый понос), нарастает интоксикация. Ребенок становится вялым, отказывается от груди или бутылочки; у него быстро прогрессируют гипотрофия и кахексия. Ребенка могут беспокоить метеоризм, кишечные колики, обильное отхождение газов.
В процессе обследования ребенка с галактоземией неонатологом выявляется угасание рефлексов периода новорожденности. При галактоземии рано появляется стойкая желтуха различной степени выраженности и гепатомегалия, прогрессирует печеночная недостаточность. К 2-3 месяцу жизни возникают спленомегалия, цирроз печени, асцит.
Нарушения процессов свертывания крови приводит к появлению кровоизлияний на коже и слизистых оболочках. Дети рано начинают отставать в психомоторном развитии, однако степень интеллектуальных нарушений при галактоземии не достигает такой тяжести, как при фенилкетонурии. К 1-2 месяцам у детей с галактоземией выявляется двусторонняя катаракта. Поражение почек при галактоземии сопровождается глюкозурией, протеинурией, гипераминоацидурией. В терминальной фазе галактоземии ребенок погибает от глубокого истощения, тяжелой печеночной недостаточности и наслоения вторичных инфекций.
При галактоземии средней тяжести также отмечается рвота, желтуха, анемия, отставание в психомоторном развитии, гепатомегалия, катаракта, гипотрофия. Галактоземия легкой степени характеризуется отказом от груди, рвотой после приема молока, задержкой речевого развития, отставанием ребенка в массе и росте. Однако даже при легком течении галактоземии продукты обмена галактозы токсическим образом воздействуют на печень, приводя к ее хроническим заболеваниям.
Галактоземия может протекать в моносимптомной форме, при которой обнаруживается только поражение ЦНС, катаракта или диспепсические расстройства. Описан вариант бессимптомной (асимптоматической) галактоземии Дюарте, при которой недостаточность ферментов выявляется только при биохимическом исследовании крови.
Осложнения галактоземии включают цирроз печени, сепсис, кровоизлияния в стекловидное тело, первичную аменорею, синдром истощения яичников. При галактоземии у 50% детей дошкольного возраста выявляется моторная алалия, характеризующаяся трудностью организации и координации речевых движений, бедностью словарного запаса, обилием парафазий и персевераций при сохранном понимании обращенной речи.
Диагностика галактоземии
Для снижения риска развития осложнений при галактоземии необходимо как можно более раннее выявление патологии. Возможна пренатальная диагностика галактоземии, включающая проведение биопсии хориона, амниоцентеза с последующим исследованием ворсин и амниотической жидкости. В России, согласно современным стандартам, осуществляется скрининг новорожденных на следующие наследственные заболевания: фенилкетонурию, врожденный гипотиреоз, галактоземию, адрено-генитальный синдром и муковисцидоз. Неонатальный скрининг проводится на 3-5 сутки у доношенных детей и 7-10 сутки – у недоношенных. С этой целью производится забор капиллярной крови, которая переносится на фильтровальную бумагу и виде высушенных пятен отправляется в генетическую лабораторию.
Если при неонатальном скрининге у ребенка выявляется подозрение на галактоземию, проводится повторное решающее тестирование. В случае повторного обнаружения высокого уровня галактозы в крови или низкого уровня исследуемого фермента, ребенку устанавливается диагноз галактоземии. Сведения о таком ребенке сообщаются участковому педиатру, а семья новорожденного приглашается на консультацию генетика в медико-генетическую консультацию. Врач-генетик проводит подробный анализ родословной, выполняет генетическое тестирование для выявления мутантного гена, объясняет специфику питания ребенка с галактоземией.
Иногда для диагностики галактоземии прибегают к определению уровня галактозы в моче, проведению нагрузочных проб с галактозой и глюкозой. Мониторинг биохимических показателей крови и общего анализа крови и мочи при галактоземии позволяет определить степень повреждения внутренних органов (почек, печени и др.).
Дети с галактоземией нуждаются в консультации детского невролога, детского офтальмолога, проведении электроэнцефалографии, УЗИ органов брюшной полости, биомикроскопии глаза. В некоторых случаях показана пункционная биопсия печени. Галактоземию следует дифференцировать от других гликогенозов, сахарного диабета I типа, врожденной атрезии желчных протоков, гепатита, гемолитической болезни новорожденных.
Лечение галактоземии
Основная роль в лечении галактоземии принадлежит диетотерапии. Особенность питания при галактоземии заключается в пожизненном исключении из рациона продуктов, содержащих лактозу и галактозу: любого молока (женского, коровьего, козьего, детских молочных смесей, низколактозных смесей и пр.), всех молочных продуктов, хлеба, выпечки, колбас, конфет, маргаринов и др. При галактоземии запрещается употребление растительных и животных продуктов, содержащих потенциальные источники галактозы — галактозиды (бобовые, соя) и нуклеопротеины (почки, печень, яйца и др.).
Дети, страдающие галактоземией, обеспечиваются специальными смесями на основе изолята соевого белка, гидролизата казеина, синтетических аминокислот, а также безлактозными казеинпредоминантными молочными смесями. С 4-х месячного возраста вводятся фруктовые и ягодные соки; с 4,5 месяцев — фруктовое пюре; с 5 месяцев – овощное пюре; с 5,5 месяцев – безмолочные каши из кукурузной, гречневой или рисовой муки в разведении специализированной смесью; с 6 месяцев — мясной прикорм на основе мяса кролика, цыпленка, индейки, говядины; с 8 месяцев – рыба. Альтернативным источником углеводов для пациентов с галактоземией служат продукты на основе фруктозы.
Для улучшения метаболических процессов назначаются поливитамины, кокарбоксилазу, АТФ, оротат калия. Лицам с галактоземией противопоказан прием спиртовых настоек и гомеопатических препаратов, поскольку последние содержат лактозу. Дети с речевыми нарушениями нуждаются в консультации логопеда и целенаправленной работе по коррекции ОНР.
Прогноз и профилактика галактоземии
Лечение галактоземии, начатое с первых дней жизни позволяет избежать развития цирроза, катаракты, олигофрении. Если лечение начато в более поздние сроки, когда уже произошло поражение печени и ЦНС, с помощью рациональной диетотерапии прогрессирование заболевания можно замедлить. При тяжелых формах галактоземии может быть летальный исход. Диспансерное наблюдение ребенка с галактоземией осуществляется педиатром, генетиком, диетологом, детским окулистом и детским неврологом. Детям с галактоземией присваивается инвалидность.
Учитывая наследственную обусловленность галактоземии, медико-генетическое консультирование рекомендуется пройти будущим родителям, в чьих семьях есть родственники или дети с данным заболеванием. Беременным с высоким риском рождения ребенка с галактоземией, следует ограничить употребление молочных продуктов.
Не передать недуг по наследству


Ольга Васильевна, есть мнение, что генетик, как и онколог, — очень эмоционально затратная медицинская специальность: у врачей часто случается профессиональное выгорание, а пациенты воспринимают направление к генетику как приговор. Так ли это?
Ольга Удалова: В основном генетические заболевания — это редкие или орфанные. Диагностика таких заболеваний формирует персонифицированное направление медицины. Орфанный диагноз подразумевает разработку индивидуального ведения, лечения и реабилитации «редкого» пациента.
Точный генетический диагноз позволяет определить прогноз потомства в семье. Современные медицинские технологии дают возможность планировать рождение здорового ребенка даже при отягощенном семейном анамнезе.

На приеме у генетика чаще всего бывают очень тяжелые пациенты с задержкой физического и психомоторного развития. При многих наследственных заболеваниях не существует патогенетической терапии (направленной на устранение или подавление механизмов развития болезни. — Прим. ред.). В этих случаях проводятся симптоматическое лечение и реабилитация. Такие меры также могут иметь эффект и обеспечивают качество жизни отдельно взятой семьи.
В плане лечения в последние годы происходят революционные изменения: появляются и разрабатываются новые методы ферментной заместительной терапии, генотерапии, трансплантации клеток и тканей.
Так ли уж редки орфанные заболевания? И кому нужно обследоваться в обязательном порядке?
Ольга Удалова: Насчитывается более восьми тысяч орфанных заболеваний. По отдельности они редкие, но в общей массе встречаются достаточно часто. Для наследственной патологии характерно поражение многих систем и органов, поэтому орфанные пациенты могут наблюдаться у различных специалистов — неврологов, гастроэнтерологов, ортопедов, педиатров, гематологов и других. С расширением диагностических возможностей в практике врача встречается все большее число редких болезней.
Генетические заболевания имеют различный тип наследования. Доминантные проявляются с высокой вероятностью возникновения из поколения в поколение. Некоторые из них дают о себе знать сразу после рождения, другие — на третьем-четвертом и даже пятом десятке жизни.
При заболеваниях с рецессивным типом наследования родители являются здоровыми носителями мутации. В этих семьях существует высокий генетический риск для потомства. Сразу после рождения у ребенка могут отсутствовать признаки наследственного заболевания. Однако с возрастом клинические симптомы нередко нарастают, приводят к критической ситуации, тяжелому заболеванию и даже гибели.

В рамках государственной программы в России проводится неонатальный скрининг. Это массовое обследование новорожденных на пять наследственных заболеваний: адреногенитальный синдром, галактоземию, врожденный гипотиреоз, муковисцидоз, фенилкетонурию. Раннее выявление и своевременное лечение позволяют предотвратить развитие болезни у ребенка.
Вы упомянули скрининг новорожденных, есть еще скрининг беременных. Некоторые пугаются этой процедуры. Для чего проводятся скрининги и как нужно воспринимать их результаты?
Ольга Удалова: Пренатальный скрининг показан каждой беременной для того, чтобы определить состояние плода. В результате скрининга выявляется (или не выявляется) риск по хромосомной патологии у ребенка. Если пациентка попала в группу риска — это не приговор и не повод для стресса, а показание для консультации генетика. На приеме врача определяется необходимость дальнейших диагностических мероприятий, которые могут включать и инвазивные процедуры.
Очень важна консультация грамотного специалиста, который сможет максимально бережно донести до семьи эту информацию. Определяющим является решение самой семьи.
Является ли возраст будущей мамы существенным фактором риска наследственных патологий у ребенка?
Ольга Удалова: У женщин старше 35 лет увеличивается риск рождения детей с синдромом Дауна и другими хромосомными заболеваниями. Поэтому генетики рекомендуют планировать деторождение до 35 лет. Но жизнь диктует свои условия. Современные женщины хотят сделать карьеру, достичь определенного уровня благополучия. Это тоже правильно. Беременной старше 35 лет показано пройти пренатальный скрининг и обоснованно подойти к необходимости инвазивной диагностики. Поэтому возраст женщины является лишь одним из факторов риска хромосомной патологии.
Поделитесь счастливыми историями ваших «редких» пациентов?
Ольга Удалова: Счастливые случаи, безусловно, есть. Эта история началась в 1991 году. Ко мне на консультативный прием пришел пациент с целью уточнения диагноза. В процессе клинического и лабораторного обследования был поставлен диагноз: болезнь Фабри. Это заболевание неуклонно прогрессирует и чревато проявлениями почечной недостаточности, ранними инсультами, инфарктами.

В то время практики лечения болезни Фабри еще не существовало. При анализе родословной мы выявили, что у пациента есть 12 братьев и сестер. Часть братьев, поскольку заболевание связано с полом, имеют сходную клиническую картину. Одна из сестер, которая жила в соседней области, была беременна. Мы связались с ней и сообщили о риске заболевания для ее сыновей. У нее родился мальчик. При генетическом исследовании у ребенка были выявлены патологии — проявления первых признаков болезни Фабри. Парень вырос и переехал жить в Нижний Новгород, с 2013 года получает ферментзаместительную терапию. Работает, неплохо себя чувствует. Он создал прекрасную семью, у него родился здоровый сын. Я считаю, что это счастливая история.
Не секрет, что многие наследственные заболевания не имеют четкой клинической картины и маскируются под другие. Поэтому выявление наследственного заболевания крайне важно. Правильность диагноза — это правильность лечения и ведения пациентов, это вопрос не только качества жизни, но и самой жизни.
На территории Кировской областной клинической психиатрической больницы имени академика Бехтерева провели благоустройство и озеленение участка возле детского отделения учреждения. Здесь оказывают психиатрическую, психотерапевтическую, логопедическую и психологическую помощь детям в возрасте от 3 до 15 лет. Отделение рассчитано на 90 коек, из них 60 предназначены для оказания помощи несовершеннолетним с речевой патологией: заиканием, моторной алалией, дислексией и другими заболеваниями.
«Создание комфортной терапевтической среды способствует эффективному лечению, усиливает социореабилитационный потенциал для пациента, его семьи и социально значимого окружения», — говорит Игорь Набатов, главврач учреждения.
Галактоземия
, MD, Sidney Kimmel Medical College of Thomas Jefferson University
Галактоза присутствует в молочных продуктах, фруктах и овощах. Аутосомно-рецессивные дефициты фермента вызывают 3 клинических синдрома.
Дефицит галактозо-1-фосфатуридилтрансферазы
Этот дефицит вызывает классическую галактоземию. Заболеваемость составляет 1/62 000 родов, частота носительства – 1/125. Дети становятся анорексичными и желтушными в течение нескольких дней или недель потребления грудного молока или продуктов, содержащих лактозу. Возникают рвота, гепатомегалия, слабый рост, вялость, понос и сепсис (обычно Escherichia coli), так же как и нарушение функции почек (например, протеинурия, аминоацидурия, синдром Фанкони), что приводит к метаболическому ацидозу и отеку. Может также возникнуть гемолитическая анемия.
Без лечения дети остаются низкорослыми, и у них развиваются нарушения когнитивной функции, речи, походки и баланса в подростковом возрасте; у многих также возникают катаракта, остеомаляции (вызванные гиперкальциурией) и преждевременное угасание функции яичников. Пациенты с вариантом Дуарте имеют гораздо более легкие проявления.
Дефицит галактокиназы
У пациентов развивается катаракта из-за образования галактитола, который осмотически повреждает волокна хрусталика; идиопатическая внутричерепная гипертензия (псевдоопухоль мозга) встречается редко. Заболеваемость 1/40 000 рождений.
Дефицит уридиндифосфат галактоза 4-эпимеразы
Выделяют доброкачественный и тяжелый фенотипы. Встречаемость доброкачественной формы составляет 1/23000 родов в Японии; нет доступных данных по возникновению более тяжелой формы. Доброкачественная форма ограничивается изменениями в эритроцитах и лейкоцитах и не вызывает каких-либо клинических отклонений. Тяжелая форма вызывает синдром, не отличимый от классической галактоземии, иногда, с потерей слуха.
Диагностика
Определение активности ферментов
Диагноз предполагают на основании клинических данных и подтверждают при выявлении повышенного уровня галактозы и присутствии в моче восстанавливающих субстанций, отличных от глюкозы (например, галактозы, галактозо-1-фосфата); это подтверждают анализом ДНК или анализом активности ферментов в эритроцитах, анализом печеночной ткани, или того и другого. В большинстве стран требуется рутинный неонатальный скрининг на дефицит галактозо-1-фосфатной уридилтрансферазы. (Также см. Исследования при подозрении наследственных нарушений обмена веществ).
Лечение
Ограничение галактозы в диете
Лечение галактоземии требует изъятия всех источников галактозы в рационе, особенно лактозы (источник галактозы), которая присутствует в грудном молоке, в том числе продуктах на основе молока для грудных детей и подсластителях, и используется во многих продуктах питания. Безлактозная диета предотвращает острое отравление и вызывает обратное развитие некоторых проявлений (например, катаракты), но не может предотвратить нейрокогнитивный дефицит. Многие пациенты нуждаются в дополнительном потреблении кальция и витаминов. Для пациентов с дефицитом эпимеразы потребление некоторого количества галактозы очень важно для обеспечения снабжения уридин-5′-дифосфат-галактозой (УДФ-галактоза) различных метаболических процессов.
Галактоземия
Галактоземия – это наследственное заболевание, которое протекает очень тяжело как у детей, так и у взрослых. При галактоземии организм больного человека не способен расщеплять галактозу. Галактоза (в свою очередь) является составной частью лактозы, которая содержится в молоке, поэтому больным галактоземией противопоказано употреблять молочные продукты.
Причины
Симптомы
Наиболее важные проявления патологии – это непереносимость любого молока, в составе которого имеется лактоза, а также развитие желтухи, катаракты (сложная болезнь глаз), гепатомегалии, цирроза, истощения ввиду нарушения пищеварения.
Симптомы галоктоземии у новорожденных развиваются из-за значительного повышения концентрации токсичных веществ в крови и появления реакции торможения ферментов на активность токсинов. Ферменты, участвующие в метаболизме углеводов, не могут снизить плотность галактозы (а также ее обменных продуктов), поэтому развивается гипогликемический синдром, оказывающий негативное влияние на ряд внутренних органов.
Распространенные признаки галактоземии. Виды патологии
Классический: обусловлен недостатком галактозо-1-фосфат уридилтрансферазы (это фермент, который участвует в преобразовании галактозы в глюкозу). При недостаточном количестве этого фермента происходит повышение галактозы в крови и уменьшение глюкозы. Печень неполноценно участвует в обмене D-галактозы (составной части молока). Симптомы галактоземии при классическом типе – это большая масса тела новорожденного ребенка. Дополнительно в ближайшее время после родов, после того, как малыш выпьет молоко, у него возникает жидкий стул и/или сильная рвота. Стремительно развивается гипотрофия (дефицит массы тела, истощение), появляется пожелтение склер глаз, кожи. Печень увеличивается, развиваются признаки катаракты (она вызвана образованием галактита в глазном хрусталике по причине расстройства электролитного баланса и повышения галактозы). Часто развивается гемолитическая форма анемии (распад эритроцитов).
Недостаточность галактокиназы проявляется только одним признаком – развитием катаракты. Чтобы понять, что у новорожденного ребенка развивается именно галактоземия, исследуются биологические жидкости. Анализ крови покажет увеличение галактозы и галактитола. Моча насыщается восстанавливающими веществами. Симптомы катаракты развиваются очень быстро, и галактоземия прогрессирует при отсутствии лечения. При своевременной терапии патологические процессы в глазах обратимы.
Недостаточность эпимеразы мало изучена. Патология этого типа почти не имеет физических проявлений, отмечается биохимическими сдвигами, выявляемыми случайно (обнаруживается повышение моносахарида). Данный тип болезни не требует экстренной терапии.
Галактоземия у новорожденных выдает симптоматику в первые 3–10 дней после рождения. В организм ребенка поступает молоко, а вместе с ним вещества, которые не могут перевариться и усвоиться. Постепенно токсины накапливаются. Метаболизм фермента протекает в печени, после чего он поступает в кровяное русло.
Степени тяжести
1. Легкая, при которой симптомы галактоземии видны лишь спустя 7–10 дней. Признаки болезни незначительны, токсическое воздействие моносахарида проявляется в нарушении функционирования печени (хронические патологии). Одним из признаков легкой формы галактоземии является непереносимость ребенком молока, отказ от груди.
При малосимптомном течении галактоземия поражает ЦНС, глаза, может беспокоить диспепсический синдром. Также известны случаи бессимптомного течения. Диагностика этой формы галактоземии состоит в проведении ферментного анализа.
2. Средняя степень наследственной патологии галактоземии проявляется у детей следующим образом:
- Нарушение пищеварения, потеря веса, рвоты;
- Печень в течение всего нескольких дней увеличивается в размерах;
- Желтый цвет кожи разной степени выраженности;
- Развитие анемии;
- Слизистая глаз меняется, появляется желтушность и помутнение хрусталика (свидетельствует о появлении катаракты);
- Ребенок довольно апатичен либо же, наоборот, перевозбужден (проявления со стороны ЦНС);
- Малыш «не успевает» догнать здоровых сверстников, со временем отставая в развитии двигательного аппарата, органов чувств.
3. Если степень болезни тяжелая, то симптомы галактоземии развиваются почти молниеносно в самые первые дни после рождения. Кормление молоком с лактозой вызывает:
- Рвоту, обычно такую обильную, что развивается стремительное истощение;
- Жидкие испражнения;
- Колики и отхождение газов;
- Интоксикация, отказ от груди (либо бутылочки);
- Свойственные новорожденным рефлексы угасают;
- Желтушность и увеличение размеров печение, поражение почек;
- В возрасте 1–2 месяца диагностируется двусторонняя катаракта;
- Симптомы галактоземии в возрасте 2–3 месяцев – это цирроз, жидкость внутри брюшной ямы, увеличение селезенки (спленомегалия);
- Кровоизлияние (на слизистых, коже – из-за нарушения свертываемости);
- Со временем развивается нарушение интеллекта;
Далее присоединяются вторичные инфекции (если нет лечения).
Катаракта
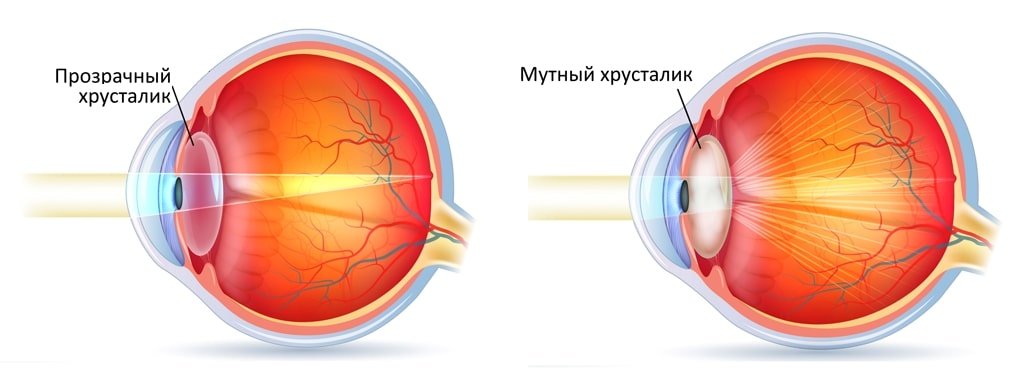
Катаракта – это частичное или полное помутнение хрусталика глаза, расположенного внутри глазного яблока между радужкой и стекловидным телом. Хрусталик от природы прозрачный и играет роль естественной линзы, преломляющей световые лучи и пропускающей их к сетчатке. Потерявший прозрачность хрусталик при катаракте перестаёт пропускать свет и зрение ухудшается вплоть до полной потери.
Причины развития катаракты и группы риска
Катаракта может развиться:
- у пожилых людей – возрастная катаракта (90% всех случаев катаракты);
- у людей после травм – травматическая катаракта (4%);
- после радиационного облучения – лучевая катаракта (3%);
- у новорождённых – врождённая катаракта (3%).
Развитию катаракты способствуют эндокринные расстройства (нарушение обмена веществ, сахарный диабет), авитаминоз, некоторые глазные заболевания, неблагоприятная экологическая обстановка, длительный приём определённых лекарственных препаратов. В последние годы было доказано, что причиной катаракты может также стать активное курение.
Симптомы катаракты
-small.jpg)
Древние греки называли катаракту «водопад» – kataraktes. Человек видит как бы сквозь пелену, запотевшее стекло. Это и есть главный симптом катаракты, говорящий о том, что помутнение уже затронуло центральную зону хрусталика и требуется оперативное лечение.
Как видит человек с катарактой








Здесь перечислены далеко не все признаки катаракты. И, конечно, важно понимать, что те же симптомы могут свидетельствовать о наличии других опасных заболеваний глаз! Определить, что именно стало причиной ухудшения зрения и принять адекватные лечебные меры под силу только грамотному специалисту.
В состав хрусталика входят соединения белка, обладающими определёнными физико-химическими и биологическими свойствам и обеспечивающими прозрачность хрусталика. Под влиянием возрастных изменений в организме происходит денатурация белковых соединений – нарушение структуры молекул белка, то есть потеря их природных свойств. Представьте себе яичный белок: при варке он теряет прозрачность и становится белым. Это и есть процесс денатурации, при этом прозрачность вернуть белку уже невозможно. Можно сказать, что аналогичные процессы происходят и в хрусталике глаза человека.
Как быстро созревает катаракта?
Процесс помутнения хрусталика необратим!
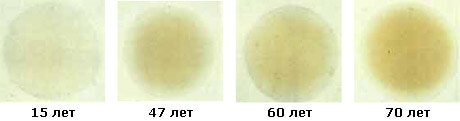
Хрусталик при возрастной катаракте мутнеет постепенно, процесс может занять от 4 до 15 лет. На начальной стадии катаракты помутнения могут затрагивать лишь периферию хрусталика, не попадать в оптическую зону и не влиять на зрение. Затем изменения охватывают и центральную часть, препятствуя прохождению света, и зрение ощутимо ухудшается. При перезрелой катаракте острота зрения снижается до светоощущения. По мере развития катаракты цвет зрачка вместо чёрного постепенно становится сероватым, серовато-белым, молочно-белым. В этих случаях катаракту можно заметить даже без специального оборудования.
Возрастные изменения хрусталика
Катаракта у пожилых людей
Чаще всего встречается возрастная катаракта. По статистике Всемирной организации здравоохранения, в 70-80% случаев катаракта развивается у людей после 70 лет. Однако возрастная катаракта может развиться и раньше, в возрасте 45 – 50 лет. Нередко возрастную катаракту называют старческой, но такое название нельзя считать корректным.
Основная причина развития возрастной катаракты – изменение биохимического состава хрусталика, обусловленное возрастными процессами в организме. Помутнение хрусталика с точки зрения функционирования организма человека – вполне естественное явление, поэтому от катаракты никто не застрахован.
Врождённая катаракта у ребенка
Врождённая катаракта составляет более половины всех врождённых дефектов органа зрения. Катаракта у новорождённых обусловлена генетическими изменениями в структуре белков, необходимых для обеспечения прозрачности хрусталика. Причинами катаракты у детей до года могут быть сахарный диабет у матери, инфекционные заболевания матери в I триместре беременности, приём определённых лекарственных препаратов. Главное в этом случае – ранняя диагностика врождённой катаракты. Если локализация и размеры помутнения в хрусталике не препятствуют правильному развитию органа зрения, то такая катаракта не требует экстренного хирургического лечения.
Среди врождённых катаракт наиболее часто встречаются:
- КАПСУЛЯРНАЯ. Изолированное помутнение передней или задней сумки (капсулы) хрусталика. Степень снижения зрения зависит от размеров помутнения капсулы. Развитие капсулярной катаракты может быть вызвано заболеваниями матери во время беременности или внутриутробными воспалительными процессами.
- ПОЛЯРНАЯ. Поражение распространяется как на капсулу, так и на вещество хрусталика у переднего или заднего полюсов. В большинстве случаев встречается двусторонняя катаракта. Размеры и форма значительно варьируются, от чего зависит ее влияние на зрение.
- СЛОИСТАЯ (зонулярная). Самая часто встречающаяся форма врожденной катаракты. В подавляющем большинстве случаев двусторонняя. Располагается в центре, вокруг прозрачного (или слегка мутноватого) ядра. Зрение снижается всегда, чаще всего очень значительно, до 0,1 и ниже.
- ЯДЕРНАЯ. Развивается на обоих глазах, имеет выраженный семейно-наследственный характер, чаще всего снижается зрение до очень низкого уровня — 0,1 и ниже. В случаях, когда помутнение ограничивается эмбриональным ядром, зрение может снижаться незначительно или не падать вовсе.
- ПОЛНАЯ. Заболевание, как правило, двустороннее. Клиническая картина разнообразна и зависит от степени помутнения хрусталика. При полном развитии катаракты весь хрусталик мутный. Ребенок слеп, имеет только светоощущение. Может развиться еще до рождения или созреть в первые месяцы жизни. Полная катаракта сочетается с другими дефектами развития глаз (микрофтальмом, колобомой сосудистой оболочки, гипоплазией желтого пятна, нистагмом, косоглазием и т. д.). Полная катаракта иногда может иметь тенденцию к рассасыванию, и тогда в области зрачка остается пленка — пленчатая катаракта.
- ОСЛОЖНЕННАЯ. Причиной ее развития может быть галактоземия, диабет, вирусная краснуха и другие тяжелые заболевания. Часто сопровождается другими врожденными дефектами (пороки сердца, глухота).
Главное в этом случае – ранняя диагностика врождённой катаракты. Если локализация и размеры помутнения в хрусталике не препятствуют правильному развитию органа зрения, то такая катаракта не требует экстренного хирургического лечения. Если же помутнение препятствует поступлению световых лучей к сетчатке, развитию центрального зрения у младенца, то необходимо как можно раньше удалить это препятствие для того, чтобы правильно развивалась зрительная система ребёнка. Лечение врождённой катаракты в клинике «Эксимер» проводится даже у самых маленьких детей, начиная с трёх месяцев.
Диагностика катаракты
Современные диагностические приборы и методики в клинике «Эксимер» позволяют выявить катаракту даже на ранних стадиях. На начальной стадии развития катаракты человек может и не подозревать о наличии заболевания, однако биохимический процесс уже запущен, хрусталик постепенно потеряет прозрачность, а человек – зрение. Многие путают катаракту с другим схожим по симптомам, но иным по своей природе возрастным изменением – возрастной дальнозоркостью. Точный диагноз поставит только врач-офтальмолог при помощи специальных приборов. Основным в диагностике катаракты является осмотр с помощью световой (щелевой) лампы – биомикроскопия глаза. Важны также такие исследования как определение остроты зрения, осмотр глазного дна, исследование полей зрения, измерение внутриглазного давления. На этапе расчёта параметров искусственного хрусталика в клинике «Эксимер» применяется уникальный прибор – оптический когерентный биометр ИОЛ-мастер Zeiss.
Единственный эффективный способ избавиться от катаракты – хирургическое лечение, в ходе которого помутневший хрусталик заменяется на прозрачный искусственный, по своим свойствам максимально приближенный к природному. В клинике «Эксимер» лечение катаракты проводится по самой передовой методике – ультразвуковой факоэмульсификации катаракты, в том числе и с фемтолазерным сопровождением.